Declarative compartmental models with Catalyst
The SIR case study builds its dynamics from a hand-written vector field, and the package hand-writes that model's Jacobian too so the stiff/auto solver stays fast and stable. A hand-written Jacobian has to be re-derived, and kept in sync, for every new compartmental model.
Catalyst.jl [8] removes that step. You declare a reaction network, and Catalyst together with ModelingToolkit.jl generate the ODE system and a symbolic Jacobian for you, kept consistent by construction.
The extension's CatalystODEParams is model-agnostic. It takes any Catalyst ReactionSystem, reads off its species and rate parameters symbolically, and asks only for a prior per species and per rate. The same type builds an SIR model, an SEIR model, or a network with a vaccinated class or a second strain. Only the reactions change. This page fits the SIR network to a real outbreak, then swaps in SEIR without touching the rest of the model to show the API is generic.
Optional extension
The Catalyst path lives in an optional package extension. It loads only when you add and import Catalyst (and ModelingToolkit) alongside ComposableTuringIDModels, which keeps the heavy symbolic stack out of the default install. The hand-coded SIRParams / SEIRParams remain the zero-latency default; the declarative path is opt-in for users building new or more complex compartmental models.
Declaring and grokking a network
We declare SIR as two reactions. Each reads like the transmission diagram, a rate constant then the species that react.
Catalyst expands these into the SIR drift and generates its Jacobian symbolically, so there is no vector field or Jacobian to write or maintain.
using ComposableTuringIDModels, Catalyst, ModelingToolkit, OrdinaryDiffEq
using Distributions, Random, Turing, LogExpFunctions, ADTypes
using CSV, DataFrames
Random.seed!(1978)
sir = @reaction_network begin
β, S + I --> 2I
γ, I --> R
endThe point of the generic path is that we can grok any network symbolically. Catalyst reads off the species and rate parameters we then attach priors to.
(species = species(sir), parameters = parameters(sir))(species = SymbolicUtils.BasicSymbolicImpl.var"typeof(BasicSymbolicImpl)"{SymbolicUtils.SymReal}[S(t), I(t), R(t)], parameters = SymbolicUtils.BasicSymbolicImpl.var"typeof(BasicSymbolicImpl)"{SymbolicUtils.SymReal}[β, γ])Building the parameter component
Loading Catalyst activates the extension that backs CatalystODEParams (the type itself is a normal, exported ComposableTuringIDModels component). We hand it the network, a solver time span, and a prior per species and per rate, as symbolic-handle ⇒ spec pairs. A spec is either a Distribution (sampled, and named after its symbol in the chain, β, γ) or a plain number (a fixed value, not sampled, here the empty recovered class R(0)). The (u0, p) sampling contract is the same as the hand-coded parameter models, so it drops straight into an ODEProcess.
We fit the classic influenza outbreak in an English boarding school [6], taking the children confined to bed each day as a proxy for the infectious compartment.
N = 763 # children in the school
datapath = joinpath(pkgdir(ComposableTuringIDModels),
"docs", "src", "case-studies", "data", "influenza_england_1978_school.csv")
influenza = CSV.read(datapath, DataFrame)
y_obs = influenza.in_bed # children confined to bed each day
ts = collect(1.0:length(y_obs)) # observation times (days)
n = length(y_obs)
sir_params = CatalystODEParams(sir;
tspan = (0.0, ts[end]),
u0_priors = [sir.S => 0.99, sir.I => Beta(2, 200), sir.R => 0.0],
p_priors = [sir.β => LogNormal(-0.5, 0.5), sir.γ => Gamma(8, 0.03125)])A note on species ordering
Catalyst sorts the species and parameters when it builds the problem, so the internal layout is generally not the order you wrote the network in. We never rely on that order. CatalystODEParams samples into symbolic symbol => value maps that remake places by name, and the sol2infs link indexes the solution symbolically with the network's own handle, so it pulls the infectious compartment out by identity rather than by a hard-coded position.
sir_process = ODEProcess(
params = sir_params,
sol2infs = sol -> sol[sir.I, :],
solver_options = Dict(:saveat => ts))Composing and fitting
From here nothing is Catalyst-specific. We scale the infectious proportion to expected counts with a population TransformObservationModel and a PoissonError, assemble with IDModel, and fit.
observation = TransformObservationModel(PoissonError(), x -> softplus.(N .* x))
model = IDModel(sir_process, observation)Use forward-mode autodiff for ODE models
The rest of these docs recommend Mooncake as the default AD backend, but ODE infection models are the exception, they sample under ForwardDiff today. Reverse-mode Mooncake-driven NUTS through the ODE solver is not yet supported for the hand-coded or the Catalyst model, a pre-existing Turing/SciMLSensitivity integration gap (tracked in issue #46) rather than anything introduced by Catalyst. We therefore pass AutoForwardDiff() to NUTS explicitly.
posterior = as_turing_model(model, y_obs, n)
chain = sample(
posterior, NUTS(0.9; adtype = AutoForwardDiff()),
MCMCThreads(), 500, 2; progress = false)┌ Warning: Only a single thread available: MCMC chains are not sampled in parallel
└ @ AbstractMCMC ~/.julia/packages/AbstractMCMC/C1aKp/src/sample.jl:544
┌ Info: Found initial step size
└ ϵ = 0.025
┌ Info: Found initial step size
└ ϵ = 0.05sample returns a FlexiChains chain. FlexiChains keys draws by their @varname, so we read parameters back by name directly, with no MCMCChains conversion. The basic reproduction number
using Statistics
β = vec(chain[@varname(β)])
γ = vec(chain[@varname(γ)])
(β = mean(β), γ = mean(γ), R0 = mean(β ./ γ))(β = 1.8862629025256354, γ = 0.47385836339818643, R0 = 3.982373277518902)Prior versus posterior
Sampling the same model with Prior gives prior draws over the transmission and recovery rates. Overlaying them on the posterior with PairPlots.jl shows how sharply the boarding-school outbreak identifies the mechanistic rates. The FlexiChains PairPlots extension takes a chain subset to a few keys with chain[[...]] directly.
using CairoMakie, PairPlots
prior_chain = sample(posterior, Prior(), 1000; progress = false)
pp_keys = [@varname(β), @varname(γ)]
pairplot(
PairPlots.Series(chain[pp_keys]; label = "posterior"),
PairPlots.Series(prior_chain[pp_keys]; label = "prior"))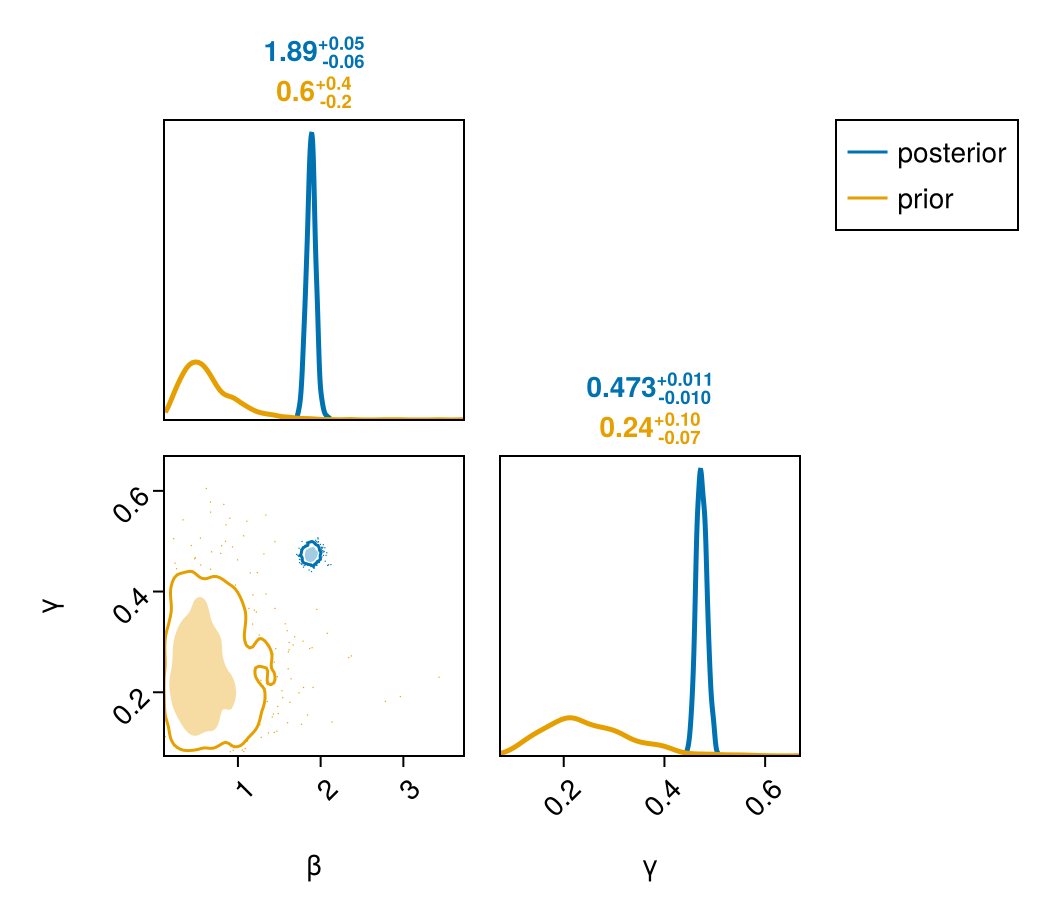
Both rates collapse from broad priors onto tight, correlated posteriors.
Posterior trajectories
A compartmental model has no time-varying nothing); the infection signal is the infectious proportion generated_observables recovers predict on the model with the observations set to missing. Two small helpers reduce the per-draw trajectories to credible bands.
gens = vec(generated_observables(posterior, y_obs, chain).generated)
It = credible_bands(reduce(hcat, (g.I_t for g in gens)))
pred = predict(as_turing_model(model, fill(missing, n), n), chain)
yt = predictive_bands(pred, n)
fig = Figure(; size = (760, 620))
ax1 = Axis(fig[1, 1]; ylabel = "Infectious proportion I(t)")
ci_ribbon!(ax1, ts, It; color = :purple, label = "posterior median")
axislegend(ax1; position = :rt)
ax2 = Axis(fig[2, 1]; xlabel = "Day", ylabel = "Children in bed")
ci_ribbon!(ax2, ts, yt; color = :teal, label = "posterior predictive")
scatter!(ax2, ts, y_obs; color = :black, markersize = 7, label = "observed")
axislegend(ax2; position = :rt)
fig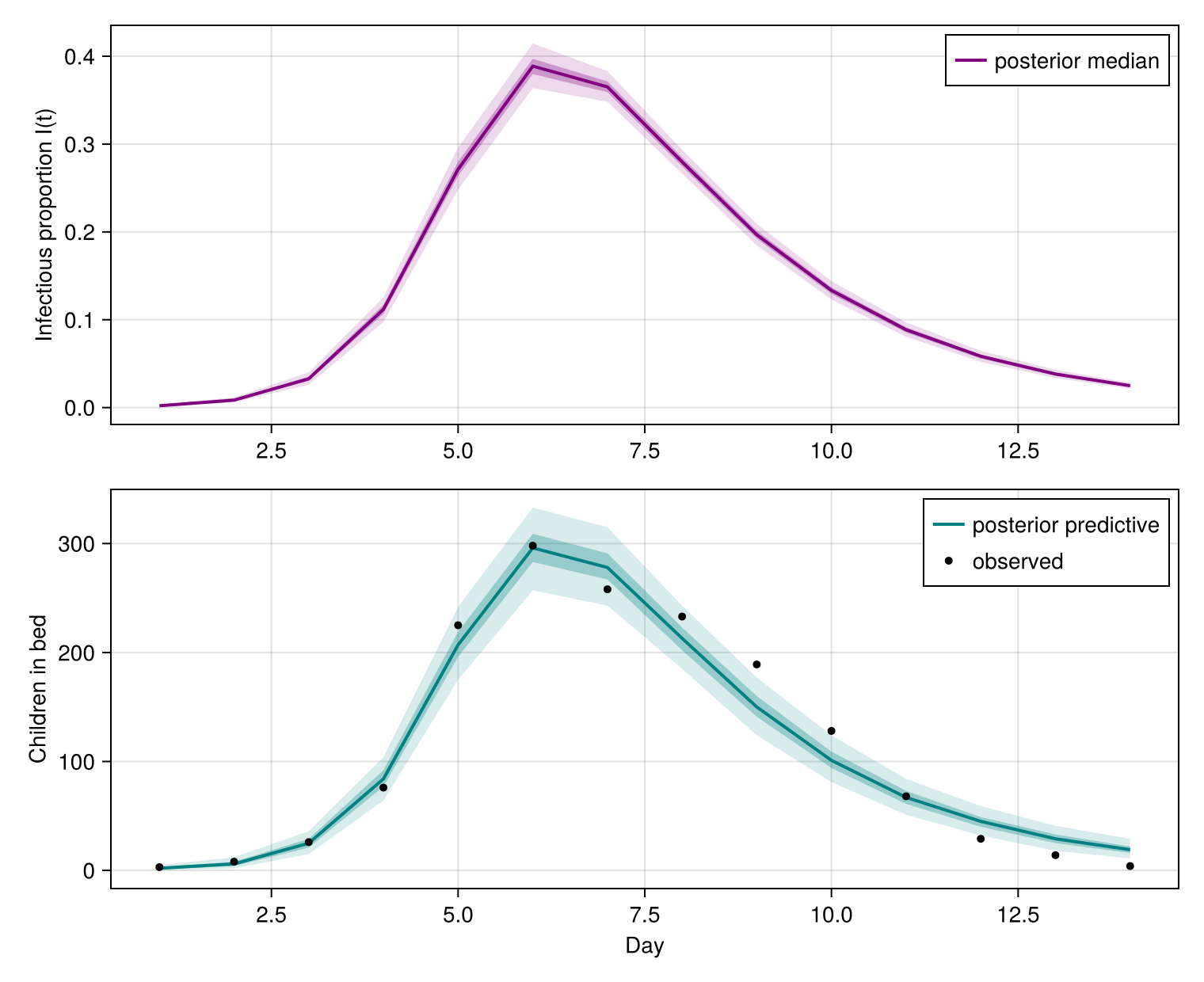
The declarative SIR dynamics, scaled by the population and Poisson observation model, reproduce the boarding-school outbreak.
The same API on a different network
The payoff of the generic path is that a different compartmental model is a different reaction network passed to the same CatalystODEParams. We add an exposed class
seir = @reaction_network begin
β, S + I --> E + I
α, E --> I
γ, I --> R
end
(species = species(seir), parameters = parameters(seir))(species = SymbolicUtils.BasicSymbolicImpl.var"typeof(BasicSymbolicImpl)"{SymbolicUtils.SymReal}[S(t), I(t), E(t), R(t)], parameters = SymbolicUtils.BasicSymbolicImpl.var"typeof(BasicSymbolicImpl)"{SymbolicUtils.SymReal}[β, α, γ])The network now has four species and three rates, and Catalyst regenerates the drift and Jacobian for us. We attach a prior per species and per rate exactly as before, index the same infectious compartment symbolically, and reuse the same observation model.
seir_params = CatalystODEParams(seir;
tspan = (0.0, ts[end]),
u0_priors = [seir.S => 0.99, seir.E => Beta(2, 200),
seir.I => Beta(2, 200), seir.R => 0.0],
p_priors = [seir.β => LogNormal(-0.5, 0.4),
seir.α => Gamma(8, 0.05), seir.γ => Gamma(8, 0.03125)])
seir_model = IDModel(
ODEProcess(params = seir_params, sol2infs = sol -> sol[seir.I, :],
solver_options = Dict(:saveat => ts)),
observation)
seir_chain = sample(
as_turing_model(seir_model, y_obs, n),
NUTS(0.9; adtype = AutoForwardDiff()), 200; progress = false)
βe = vec(seir_chain[@varname(β)])
αe = vec(seir_chain[@varname(α)])
γe = vec(seir_chain[@varname(γ)])
(β = mean(βe), α = mean(αe), γ = mean(γe), R0 = mean(βe ./ γe))(β = 3.9085902045292276, α = 1.0453668180862825, γ = 0.48993068236871534, R0 = 7.9823804044254265)Both fits ran through the same CatalystODEParams, the same ODEProcess contract, and the same FlexiChains readback. Adding a fourth compartment, a vaccinated class, or a second strain is a matter of writing a different reaction network and passing it to the same type, and the vector field and Jacobian follow automatically. That is the trade the Catalyst extension offers, a one-off symbolic-compilation cost and a heavier dependency tree in exchange for declarative, model-agnostic, self-consistent dynamics.
References
A. Chatzilena, E. van Leeuwen, O. Ratmann, M. Baguelin and N. Demiris. Contemporary statistical inference for infectious disease models using Stan. Epidemics 29, 100367 (2019).
T. E. Loman, Y. Ma, V. Ilin, S. Gowda, N. Korsbo, N. Yewale, C. Rackauckas and S. A. Isaacson. Catalyst: Fast and flexible modeling of reaction networks. PLoS Computational Biology 19, e1011530 (2023).