An SIR compartmental model
The renewal equation is one way to generate infections, but it is not the only one. Mechanistic compartmental models describe transmission with a system of ordinary differential equations (ODEs). Chatzilena et al. [6] showed how to embed such an ODE in a Bayesian model and infer its parameters, using a classic influenza outbreak in an English boarding school as their example.
This case study swaps the renewal infection process for an ODEProcess built from SIRParams, keeping the same composable observation machinery. Infections come from solving the SIR equations with the SciML stack [5] rather than from a bespoke solver.
The model
The infection process
SIRParams declares priors for the transmission rate, recovery rate, and initial infected proportion, over a solver time span. We use weakly informative priors that keep the basic reproduction number
using ComposableTuringIDModels, Distributions, Random, Turing, LogExpFunctions
using ADTypes: AutoForwardDiff
using CSV, DataFrames
Random.seed!(1978)
N = 763 # children in the school
datapath = joinpath(pkgdir(ComposableTuringIDModels),
"docs", "src", "case-studies", "data", "influenza_england_1978_school.csv")
influenza = CSV.read(datapath, DataFrame)
y_obs = influenza.in_bed # children confined to bed each day
ts = collect(1.0:length(y_obs)) # observation times (days)
n = length(y_obs)
sir_params = SIRParams(
tspan = (0.0, ts[end]),
infectiousness = LogNormal(-0.5, 0.5),
recovery_rate = Gamma(8, 0.03125),
initial_prop_infected = Beta(2, 200))Chatzilena et al. [6] fit this to a 1978 influenza outbreak in an English boarding school, taking the number of children "in bed" each day as a proxy for the infected compartment. Of the 763 children, 512 fell ill over 14 days.
ODEProcess composes those parameters with a solver and a sol2infs link that pulls the infected compartment out of the ODE solution. This is the standard SciML pattern — a problem definition composed with a solution method — specialised to probabilistically sampled parameters. The default solver switches between explicit and implicit methods, which keeps the solve robust when the sampler proposes stiff parameter values.
sir_process = ODEProcess(
params = sir_params,
sol2infs = sol -> sol[2, :],
solver_options = Dict(:saveat => ts))The observation model
The ODE returns the infected proportion TransformObservationModel, then link to data with a PoissonError.
observation = TransformObservationModel(PoissonError(), x -> softplus.(N .* x))A compartmental model needs no time-varying latent ODEProcess carries no latent process at all (its Z_t generated quantity is nothing). IDModel assembles the infection and observation parts exactly as in the renewal examples.
model = IDModel(sir_process, observation)Fit
Fitting recovers the SIR parameters from the observed "in bed" counts. This page differentiates with ForwardDiff, not the package's recommended Mooncake default: reverse-mode (Mooncake-driven) NUTS through the ODE solver is not available yet — a pre-existing Turing + Mooncake + SciMLSensitivity integration gap that affects every ODE infection model (tracked in issue #46). Forward-mode autodiff is a good fit here anyway, for a system this small. We draw two chains in parallel with MCMCThreads() so a cross-chain
posterior = as_turing_model(model, y_obs, n)
chain = sample(
posterior, NUTS(0.9; adtype = AutoForwardDiff()),
MCMCThreads(), 500, 2; progress = false)┌ Warning: Only a single thread available: MCMC chains are not sampled in parallel
└ @ AbstractMCMC ~/.julia/packages/AbstractMCMC/C1aKp/src/sample.jl:544
┌ Info: Found initial step size
└ ϵ = 0.003125
┌ Info: Found initial step size
└ ϵ = 0.05
┌ Warning: Verbosity toggle: dt_epsilon
│ Initial timestep too small (near machine epsilon), using default: dt = 1.0e-6
└ @ OrdinaryDiffEqCore ~/.julia/packages/OrdinaryDiffEqCore/it3lY/src/initdt.jl:214sample returns a FlexiChains chain, which summarystats summarises directly — no conversion step:
using MCMCChains
summarystats(chain)╭─FlexiSummary (9 statistics) ─────────────────────────────────────────────────╮
│ iter collapsed │
│ chain collapsed │
│ ↓ stat = [mean, std, mcse, ess_bulk, ess_tail, rhat, q5, q50, q95] │
│ │
│ Parameters (3) ── AbstractPPL.VarName │
│ Float64 β, γ, I₀ │
│ │
│ Extras (14) │
│ Float64 n_steps, is_accept, acceptance_rate, log_density, │
│ hamiltonian_energy, hamiltonian_energy_error, │
│ max_hamiltonian_energy_error, tree_depth, numerical_error, │
│ step_size, nom_step_size, logprior, loglikelihood, logjoint │
│ │
│ Summary │
│ param mean std mcse ess_bulk ess_tail rhat q5 … │
│ β 1.8666 0.0540 0.0031 301.6835 448.5876 0.9991 1.7762 … │
│ γ 0.4787 0.0117 0.0005 498.5997 370.6391 1.0039 0.4589 … │
│ I₀ 0.0006 0.0001 0.0000 282.8139 450.5228 0.9994 0.0004 … │
╰──────────────────────────────────────────────────────────────────────────────╯The posterior gives the transmission and recovery rates directly, and the basic reproduction number vec(chain[@varname(...)]), from which the derived
using Turing: @varname
using Statistics
β = vec(chain[@varname(β)])
γ = vec(chain[@varname(γ)])
R0 = β ./ γ
(β = mean(β), γ = mean(γ), R0 = mean(R0))(β = 1.8665869637031365, γ = 0.4786799432446836, R0 = 3.9014231771745913)Prior versus posterior
Sampling the same model with Prior gives a prior draw over the transmission rate
using CairoMakie, PairPlots
prior_chain = sample(posterior, Prior(), 1000; progress = false)
pp_keys = [@varname(β), @varname(γ), @varname(I₀)]
pairplot(
PairPlots.Series(chain[pp_keys]; label = "posterior"),
PairPlots.Series(prior_chain[pp_keys]; label = "prior"))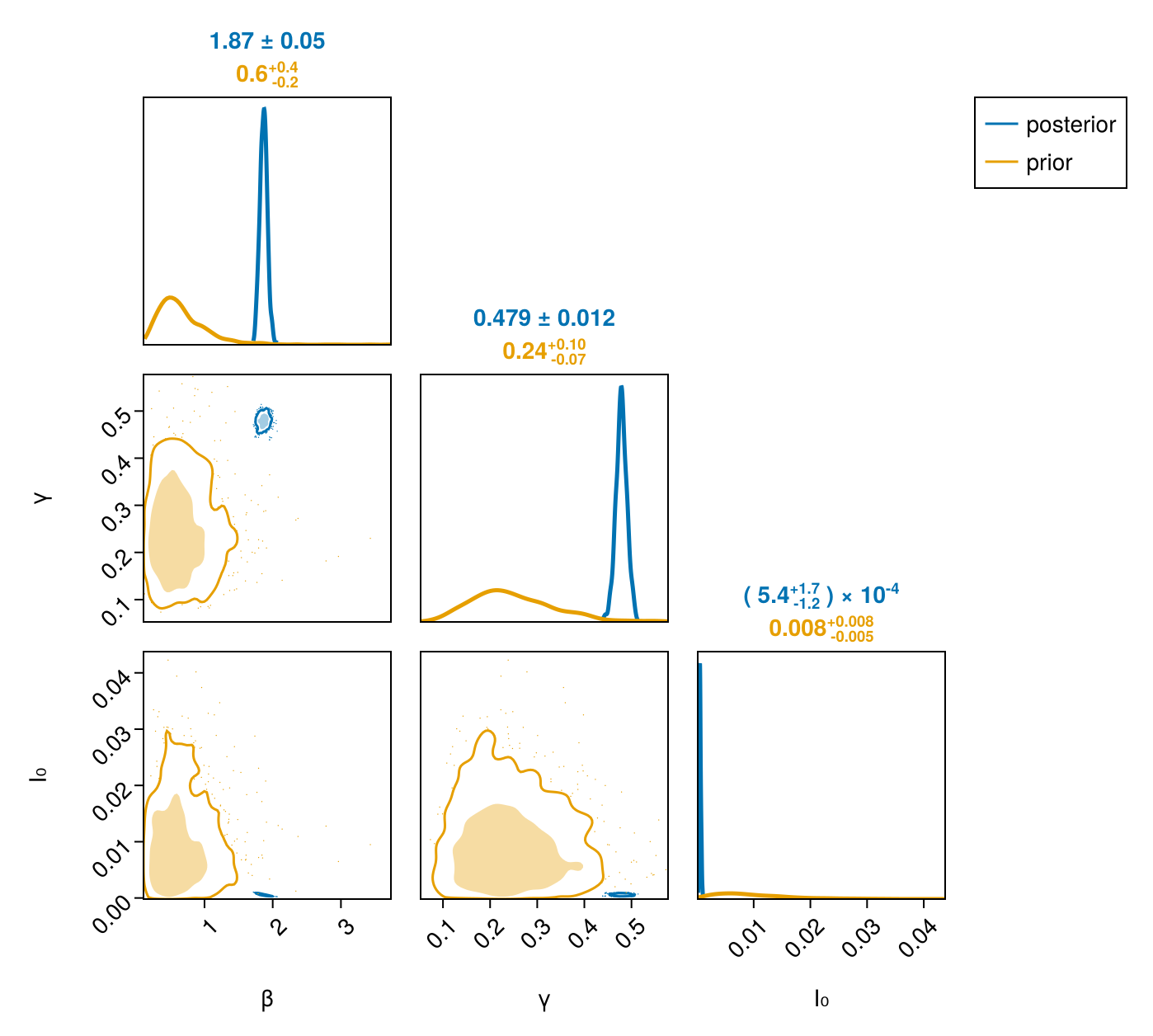
All three parameters collapse from broad priors onto tight, correlated posteriors —
Posterior trajectories
A compartmental model has no time-varying nothing); the infection signal is the infectious proportion generated_observables recovers predict on the model with the observations set to missing. Two small helpers reduce the per-draw trajectories to credible bands.
gens = vec(generated_observables(posterior, y_obs, chain).generated)
It = credible_bands(reduce(hcat, (g.I_t for g in gens)))
pred = predict(as_turing_model(model, fill(missing, n), n), chain)
yt = predictive_bands(pred, n)
fig = Figure(; size = (760, 620))
ax1 = Axis(fig[1, 1]; ylabel = "Infectious proportion I(t)")
ci_ribbon!(ax1, ts, It; color = :purple, label = "posterior median")
axislegend(ax1; position = :rt)
ax2 = Axis(fig[2, 1]; xlabel = "Day", ylabel = "Children in bed")
ci_ribbon!(ax2, ts, yt; color = :teal, label = "posterior predictive")
scatter!(ax2, ts, y_obs; color = :black, markersize = 7, label = "observed")
axislegend(ax2; position = :rt)
fig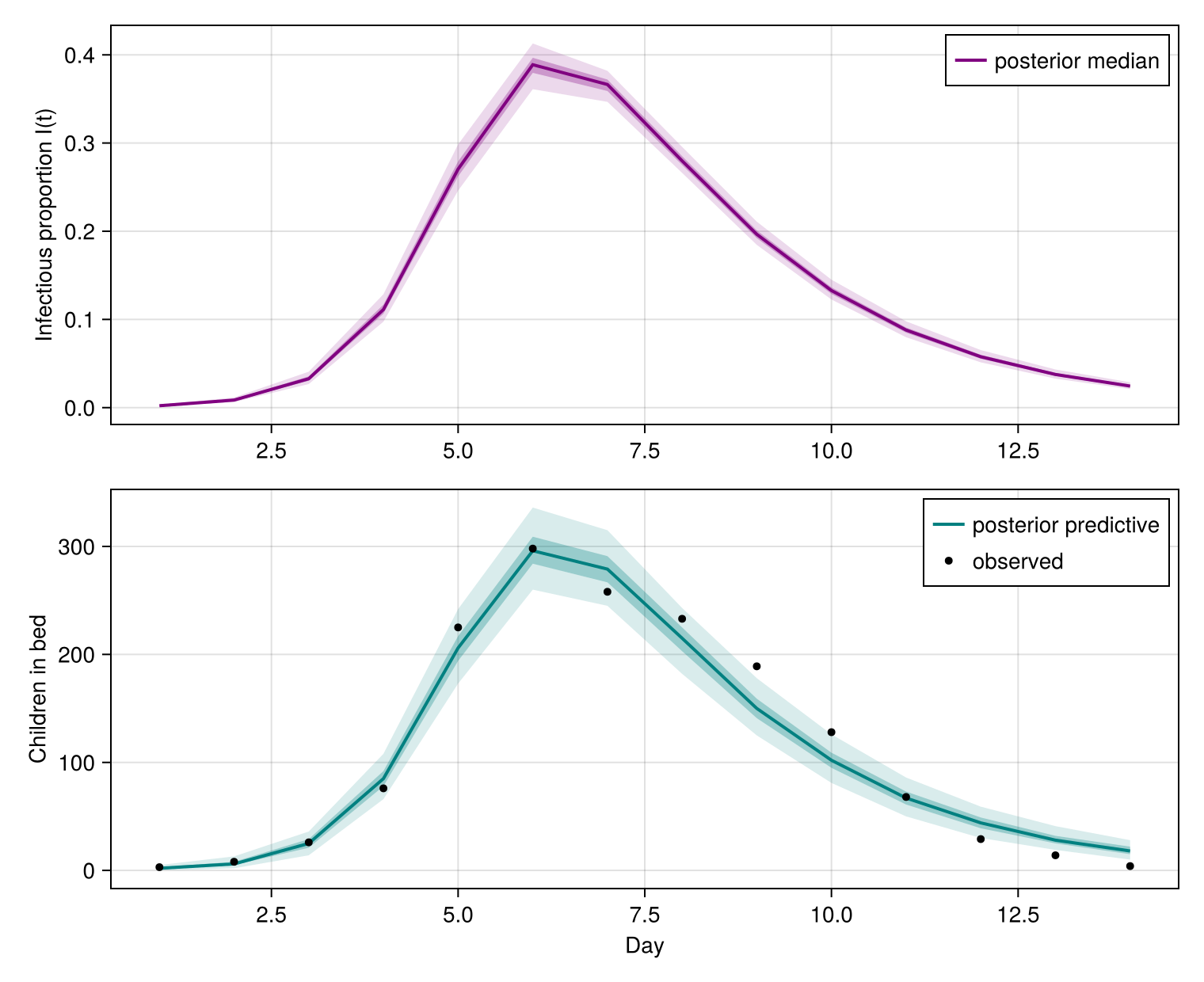
The mechanistic infectious-proportion curve peaks mid-outbreak, and the posterior-predictive in-bed counts bracket the observed epidemic curve — the SIR dynamics, scaled by the population and Poisson observation model, reproduce the boarding-school outbreak.
Adding a stochastic ascertainment process
The deterministic model assumes the SIR equations describe the data exactly up to Poisson counting noise. Real outbreaks rarely oblige: the compartmental model is an approximation, and reporting intensity drifts over time. Chatzilena et al. [6] therefore also consider a stochastic variant in which a latent autoregressive process on the log scale modulates the expected counts, absorbing variation the mechanistic part cannot explain:
Setting AR latent model already used for Ascertainment modifier wraps the Poisson link and carries that latent process; the population TransformObservationModel is re-applied on the outside. No part of the infection model changes. The priors are weakly informative: damping near zero (highly autocorrelated increments), an initial state near zero (no baseline adjustment), and a small innovation standard deviation.
ascertainment = AR(
damp_priors = [HalfNormal(0.005)],
init_priors = [Normal(0, 0.001)],
ϵ_t = HierarchicalNormal(std_prior = HalfNormal(0.02)))
stochastic_obs = TransformObservationModel(
Ascertainment(model = PoissonError(), latent_model = ascertainment),
x -> softplus.(N .* x))
stochastic_model = IDModel(sir_process, stochastic_obs)Swapping the deterministic observation model for the stochastic one is a single structural change — the SIR infection process is reused untouched — and the composed model is fit exactly as before. The ascertainment process adds latent parameters, so we raise the NUTS target acceptance rate a little to keep the sampler stable through the ODE solve.
stochastic_chain = sample(
as_turing_model(stochastic_model, y_obs, n),
NUTS(0.9; adtype = AutoForwardDiff()),
MCMCThreads(), 500, 2; progress = false)┌ Warning: Only a single thread available: MCMC chains are not sampled in parallel
└ @ AbstractMCMC ~/.julia/packages/AbstractMCMC/C1aKp/src/sample.jl:544
┌ Info: Found initial step size
└ ϵ = 0.0015625
┌ Info: Found initial step size
└ ϵ = 0.0015625The SIR parameters keep their flat names (β, γ, I₀); the ascertainment process contributes its own block, prefixed Ascertainment. because modifiers that introduce a named sub-process prefix their variables to keep them distinct. summarystats shows both blocks, including the ascertainment innovation scale Ascertainment.std), which quantifies how much observation-level noise the latent process absorbed:
summarystats(stochastic_chain)╭─FlexiSummary (9 statistics) ─────────────────────────────────────────────────╮
│ iter collapsed │
│ chain collapsed │
│ ↓ stat = [mean, std, mcse, ess_bulk, ess_tail, rhat, q5, q50, q95] │
│ │
│ Parameters (19) ── AbstractPPL.VarName │
│ Float64 β, γ, I₀, Ascertainment.ar_init[1], Ascertainment.damp_AR[1], │
│ Ascertainment.std, Ascertainment.ϵ_t[1], Ascertainment.ϵ_t[2], │
│ Ascertainment.ϵ_t[3], Ascertainment.ϵ_t[4], Ascertainment.ϵ_t[5], │
│ Ascertainment.ϵ_t[6], Ascertainment.ϵ_t[7], Ascertainment.ϵ_t[8], │
│ Ascertainment.ϵ_t[9], Ascertainment.ϵ_t[10], │
│ Ascertainment.ϵ_t[11], Ascertainment.ϵ_t[12], │
│ Ascertainment.ϵ_t[13] │
│ │
│ Extras (14) │
│ Float64 n_steps, is_accept, acceptance_rate, log_density, │
│ hamiltonian_energy, hamiltonian_energy_error, │
│ max_hamiltonian_energy_error, tree_depth, numerical_error, │
│ step_size, nom_step_size, logprior, loglikelihood, logjoint │
│ │
│ Summary │
│ param mean std mcse ess_bulk ess_tail rhat … │
│ β 1.8690 0.0628 0.0025 633.1679 530.0836 1.0001 … │
│ γ 0.4844 0.0140 0.0005 731.6800 715.0619 1.0004 … │
│ I₀ 0.0006 0.0002 0.0000 583.1143 637.5481 0.9993 … │
│ Ascertainme… -0.0000 0.0010 0.0000 848.3254 528.4041 1.0011 … │
│ Ascertainme… 0.0049 0.0037 0.0001 886.3054 462.6694 1.0006 … │
│ Ascertainme… 0.0479 0.0246 0.0012 412.4352 461.9981 0.9995 … │
│ Ascertainme… 0.0452 1.0030 0.0222 2044.1810 636.9041 1.0034 … │
│ Ascertainme… 0.0439 0.9564 0.0218 1939.1820 818.2163 0.9997 … │
│ Ascertainme… -0.3409 0.9263 0.0294 1007.0582 663.5641 1.0037 … │
│ Ascertainme… 0.5648 0.9294 0.0283 1084.6202 656.5749 1.0052 … │
│ Ascertainme… 0.1117 0.8821 0.0258 1182.0905 760.7764 1.0016 … │
│ Ascertainme… -0.3967 0.7874 0.0261 916.8044 621.6724 1.0031 … │
│ Ascertainme… 0.6212 0.8809 0.0266 1090.5511 657.0042 1.0012 … │
│ Ascertainme… 1.2646 1.0390 0.0453 554.2687 450.8532 0.9999 … │
│ Ascertainme… 1.0095 0.9716 0.0348 789.7901 694.2048 1.0123 … │
│ Ascertainme… 0.0920 0.9283 0.0221 1751.2682 669.7853 1.0069 … │
│ Ascertainme… -0.5772 0.9710 0.0238 1704.7711 662.2618 0.9999 … │
│ Ascertainme… -0.5967 1.0210 0.0291 1259.6689 647.0574 0.9992 … │
│ Ascertainme… -0.6348 1.0445 0.0276 1434.6643 716.6565 0.9998 … │
╰──────────────────────────────────────────────────────────────────────────────╯The basic reproduction number is recovered as before — a derived quantity formed per draw from the sampled
βs = vec(stochastic_chain[@varname(β)])
γs = vec(stochastic_chain[@varname(γ)])
(R0 = mean(βs ./ γs),
ascertainment_sigma = mean(vec(stochastic_chain[@varname(Ascertainment.std)])))(R0 = 3.8613219919462423, ascertainment_sigma = 0.04785462150983308)Because the deterministic model is the
(deterministic_R0 = mean(R0), stochastic_R0 = mean(βs ./ γs))(deterministic_R0 = 3.9014231771745913, stochastic_R0 = 3.8613219919462423)The SIR model is an approximation to the real transmission dynamics, so here the stochastic ascertainment process soaks up systematic departures from the SIR mean, guarding the mechanistic
References
C. Rackauckas and Q. Nie. DifferentialEquations.jl – a performant and feature-rich ecosystem for solving differential equations in Julia. Journal of Open Research Software 5 (2017).
A. Chatzilena, E. van Leeuwen, O. Ratmann, M. Baguelin and N. Demiris. Contemporary statistical inference for infectious disease models using Stan. Epidemics 29, 100367 (2019).